
Young Investigator, l'institut du thorax INSERM, France
BIOGRAPHY

AUG 30, 2016 8:00 AM PDT
From urine to the study of metabolic disease - A patient-driven strategy to decipher PCSK9 roles and functions.
Presented at:
4th Annual 24 Hours of Stem Cells™ virtual event
Speaker
Abstract
In the last 10 years, PCSK9 emerged as a promising target for the treatment of autosomal dominant hypercholesterolemia (ADH). With the emergence of induced pluripotent stem (hiPS) cells and following differentiation protocols as a model for human pathology studies, we recently demonstrated the modeling of ADH due to mutations in the LDLR gene. Our strategy to better understand the role of PCSK9 included reprogramming somatic cells from patients carrying the S127R mutation into hiPS cells, differentiate these cells into hepatocytes and characterize the intracellular impact of the mutated form of PCSK9.
To facilitate our strategy, we developed the isolation, culture, amplification and reprogramming of progenitor cells derived from urine samples (UCell). After reprogramming the hiPS cells (UhiPS), showed similar characteristics to hiPS cells classically reprogrammed from skin fibroblast. Through differentiation of the UhiPS cells hepatocytes were obtained that secrete PCSK9, internalized LDL particles and respond positively to statin treatments.To validate our model, in addition to generating hiPS cells carrying the PCSK9-GOF S127R, we reprogrammed cells from a patient carrying the PCSK9-LOF R104C/V114A and showed a spontaneously low level of LDL-cholesterol. Compared to control cells, UhiPS-S127R and –R104C/V114A differentiated in a comparable manner toward hepatocytes. In our hands, while hepatocytes carrying the S127R mutation showed a lower ability to uptake LDL, hepatocytes carrying the R104C/V114A mutations displayed the inverse, when compared to control cells. In addition, cells derived from the hypercholesterolemic patient responded positively to statin treatments, at a level comparable to the clinical response of patients carrying the same S127R mutation. Altogether, we demonstrated that differentiated hiPS cells are a relevant model to decipher PCSK9 functions and patient-derived urine samples represent a convenient source of somatic cells for such study.
To facilitate our strategy, we developed the isolation, culture, amplification and reprogramming of progenitor cells derived from urine samples (UCell). After reprogramming the hiPS cells (UhiPS), showed similar characteristics to hiPS cells classically reprogrammed from skin fibroblast. Through differentiation of the UhiPS cells hepatocytes were obtained that secrete PCSK9, internalized LDL particles and respond positively to statin treatments.To validate our model, in addition to generating hiPS cells carrying the PCSK9-GOF S127R, we reprogrammed cells from a patient carrying the PCSK9-LOF R104C/V114A and showed a spontaneously low level of LDL-cholesterol. Compared to control cells, UhiPS-S127R and –R104C/V114A differentiated in a comparable manner toward hepatocytes. In our hands, while hepatocytes carrying the S127R mutation showed a lower ability to uptake LDL, hepatocytes carrying the R104C/V114A mutations displayed the inverse, when compared to control cells. In addition, cells derived from the hypercholesterolemic patient responded positively to statin treatments, at a level comparable to the clinical response of patients carrying the same S127R mutation. Altogether, we demonstrated that differentiated hiPS cells are a relevant model to decipher PCSK9 functions and patient-derived urine samples represent a convenient source of somatic cells for such study.
You May Also Like
MAR 26, 2024 | 7:00 PM
C.E. CREDITS

The implementation of a preemptive pharmacogenomics (PGx) program in a hospital setting requires a multidisciplinary approach to ensure seamless integration of each stage of the process for...
MAR 26, 2024 | 8:00 AM
C.E. CREDITS
The implementation of a preemptive pharmacogenomics (PGx) program in a hospital setting requires a multidisciplinary approach to ensure seamless integration of each stage of the process for...
DEC 07, 2023 | 8:00 AM

As the vast landscape of genetic oncology continues to expand, the ability to understand and utilize the full potential of this rich data becomes increasingly challenging. As a result, resea...
OCT 31, 2023 | 9:00 AM

Gene therapy holds potential for treating neurological diseases by delivering genetic information into specific cell types. However, selective and efficient targeting of cell types remains c...
FEB 08, 2024 | 10:00 AM
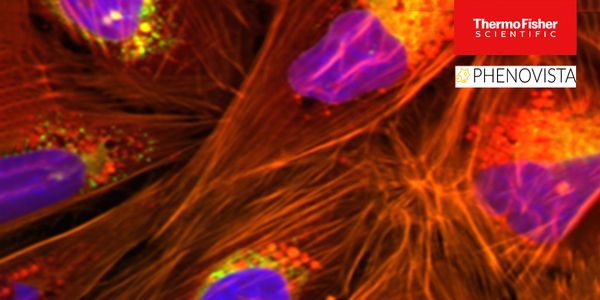
High-content screening (HCS) is an imaging-based, multi-parametric strategy used in drug development that generates rich datasets through multiplexing strategically chosen fluorescent dyes a...
NOV 08, 2023 | 8:00 AM
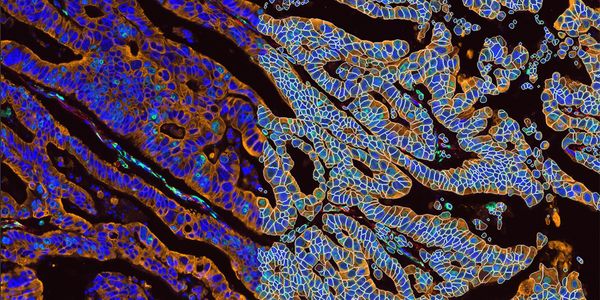
Generating Insights into tissue microenvironments is crucial to our understanding of normal and abnormal tissue development such as during cancer progression. Spatial biology methods using m...
Loading Comments...
Finish Registering
-
 APR 30, 2024Immuno-Oncology Virtual Event Series 2024
APR 30, 2024Immuno-Oncology Virtual Event Series 2024 -
MAY 07, 20243rd International Biosecurity Virtual Symposium
-
JUN 06, 2024The Future of Scientific Conferencing
- See More






-
APR 24, 2024
- See More
































